Inference of Selection from Genetic Time Series Using Various Parametric Approximations to the Wright-Fisher Model
 Selection parameter estimates in an experimental selection experiment
Selection parameter estimates in an experimental selection experimentAbstract
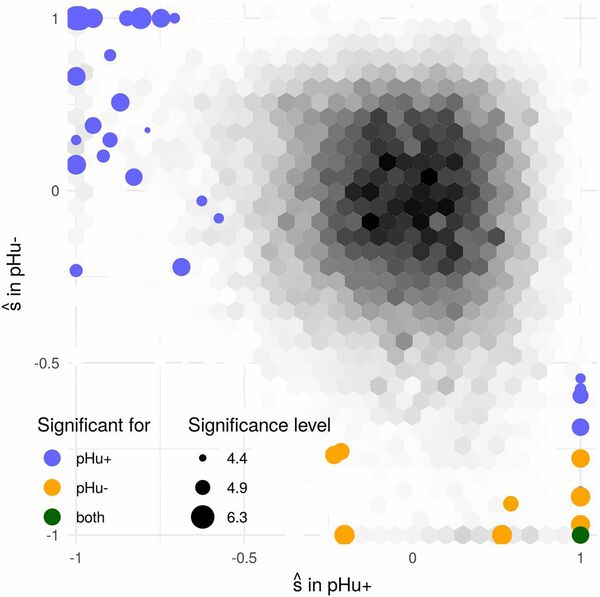
Detecting genomic regions under selection is an important objective of population genetics. Typical analyses for this goal are based on exploiting genetic diversity patterns in present time data but rapid advances in DNA sequencing have increased the availability of time series genomic data. A common approach to analyze such data is to model the temporal evolution of an allele frequency as a Markov chain. Based on this principle, several methods have been proposed to infer selection intensity. One of their differences lies in how they model the transition probabilities of the Markov chain. Using the Wright-Fisher model is a natural choice but its computational cost is prohibitive for large population sizes so approximations to this model based on parametric distributions have been proposed. Here, we compared the performance of some of these approximations with respect to their power to detect selection and their estimation of the selection coefficient. We developped a new generic Hidden Markov Model likelihood calculator and applied it on genetic time series simulated under various evolutionary scenarios. The Beta with spikes approximation, which combines discrete fixation probabilities with a continuous Beta distribution, was found to perform consistently better than the others. This distribution provides an almost perfect fit to the Wright-Fisher model in terms of selection inference, for a computational cost that does not increase with population size. We further evaluated this model for population sizes not accessible to the Wright-Fisher model and illustrated its performance on a dataset of two divergently selected chicken populations.