Imputation-Based Analysis of Association Studies: Candidate Regions and Quantitative Traits
Abstract
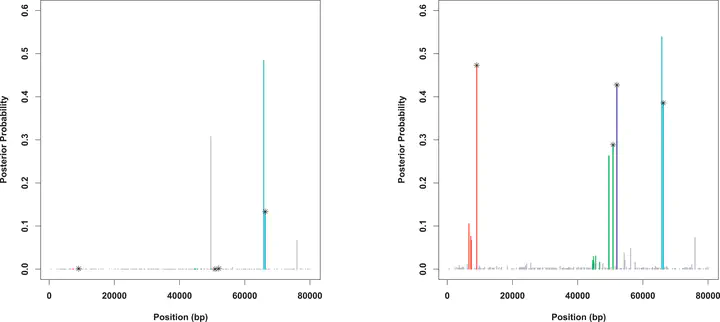
We introduce a new framework for the analysis of association studies, designed to allow untyped variants to be more effectively and directly tested for association with a phenotype. The idea is to combine knowledge on patterns of correlation among SNPs (e.g., from the International HapMap project or resequencing data in a candidate region of interest) with genotype data at tag SNPs collected on a phenotyped study sample, to estimate (“impute”) unmeasured genotypes, and then assess association between the phenotype and these estimated genotypes. Compared with standard single-SNP tests, this approach results in increased power to detect association, even in cases in which the causal variant is typed, with the greatest gain occurring when multiple causal variants are present. It also provides more interpretable explanations for observed associations, including assessing, for each SNP, the strength of the evidence that it (rather than another correlated SNP) is causal. Although we focus on association studies with quantitative phenotype and a relatively restricted region (e.g., a candidate gene), the framework is applicable and computationally practical for whole genome association studies. Methods described here are implemented in a software package, Bim-Bam, available from the Stephens Lab website http://stephenslab.uchicago.edu/software.html.